Abstract
Keywords: <em>Antiphospholipid syndrome</em>; platelet aggregation; integrin αIIbβ3; RGD-functionalized hydrogel; PEGDA; fibrin clot
Antiphospholipid syndrome (APS) is an autoimmune thrombophilia characterized by the presence of antiphospholipid antibodies (aPL), which promote vascular thrombosis and pregnancy morbidity [1–3]. In pregnant individuals, aPL can cross-react with anionic phospholipid-binding proteins on platelets and trophoblasts, resulting in hypercoagulability, inflammatory responses, and placental insufficiency [4,5]. These pathophysiological changes underlie a spectrum of obstetric complications, including recurrent miscarriage, preeclampsia, and intrauterine growth restriction [6,7].
A central mediator of aPL-induced β αIIbβ3, a platelet surface receptor that facilitates platelet–platelet aggregation via binding to fibrinogen and RGD-containing ligands [8–10]. Upon activation by aPL or inflammatory signals, αIIbβ3 undergoes a conformational change that enables strong adhesive interactions, promoting thrombus formation in the placental vasculature [11,12]. This aggregation not only contributes to local ischemia but also triggers the release of platelet-derived cytokines, which further impair trophoblast function and migration—processes critical for spiral artery remodeling and placental development [13,14].
Although current treatments such as heparin and aspirin provide partial protection, they are associated with systemic bleeding risks and do not directly target the molecular mechanisms of platelet activation [15–17]. Therefore, new strategies that locally modulate platelet–integrin interactions may offer safer and more targeted therapeutic options [18].
In this study, we propose a biomaterial-based approach to interfere with aPL-induced platelet aggregation by targeting integrin αIIbβ3 [19,20]. Specifically, we engineered PEGDA hydrogels functionalized with acrylated RGD peptides to competitively bind αIIbβ3, thereby blocking platelet–platelet adhesion [21,22]. In parallel, we evaluated the efficacy of αIIbβ3-neutralizing antibodies as a molecular benchmark. We hypothesized that RGD-functionalized hydrogels would competitively inhibit integrin αIIbβ3 and reduce antiphospholipid antibody–driven effects to a degree comparable to αIIbβ3-neutralizing antibodies. To test this, we conducted a series of in vitro assays to assess integrin binding, platelet aggregation, fibrin clot formation, trophoblast inflammatory responses, and cell migration under challenge. Our findings highlight the therapeutic potential of integrin-targeting biomaterials in preventing placental thrombosis and preserving trophoblast function in APS-associated pregnancy complications.
PEGDA (Mn ~700 Da, cat. no. 455008, Sigma-Aldrich, St. Louis, MO, USA), Irgacure 2959 (cat. no. I2909, Sigma-Aldrich, St. Louis, MO, USA), and human fibrinogen (cat. no. F3879, Sigma-Aldrich, St. Louis, MO, USA) were used. Acrylated RGD peptide (Ac-GRGDSC, >95% purity) was custom synthesized (GL Biochem, Shanghai, China). Recombinant human integrin αIIbβ3 protein (His-tag, cat. no. 3045-AB, R&D Systems, Minneapolis, MN, USA) and ELISA kits for IL-6 (cat. no. D6050) and TNF-α (cat. no. DTA00D) were purchased from R&D Systems. Human trophoblast HTR-8/SVneo cells (cat. no. CRL-3271) were obtained from ATCC (Manassas, VA, USA). Platelets were obtained from a commercial supplier (Shanghai Yuanye Bio-Technology Co., Ltd., Shanghai, China; catalog no. HPL01), which provides fully consented human platelet preparations for research purposes.
2.2. Preparation of RGD-functionalized PEGDA hydrogels and integrin binding assay
Polyethylene glycol diacrylate (PEGDA, Mn ~700 Da, Sigma-Aldrich, St. Louis, MO, USA; cat. no. 455008) was dissolved in sterile phosphate-buffered saline (PBS, pH 7.4, Gibco, Grand Island, NY, USA) to prepare a 20% (w/v) prepolymer solution. Irgacure 2959 (0.05% w/v, Sigma-Aldrich, St. Louis, MO, USA; cat. no. I2909) was added as the photoinitiator, and acrylated RGD peptide (Ac-GRGDSC, >95% purity, GL Biochem, Shanghai, China) was incorporated at final concentrations of 0, 10, 50, or 100 μmol/g. The precursor solution was sterilized by filtration through a 0.22 μm syringe filter (Millipore, Burlington, MA, USA) and cast into PDMS molds (6 mm diameter, 1 mm thickness) placed in 96-well plates. Photopolymerization was carried out under UV light (365 nm, 5 mW/cm2) for 3 min at room temperature. After curing, hydrogels were carefully removed from the molds and washed in PBS for 24 h, with solution changes every 6 h, to remove unreacted residues.
Recombinant integrin αIIbβ3 (1 μg/mL) was incubated with the hydrogels at 37°C for 1 h. After washing, anti-His HRP-conjugated antibody was applied (1:1000, 1 h), followed by TMB substrate. OD450 was measured to quantify binding.
2.3. Platelet aggregation assay under aPL stimulation
Washed platelets were prepared by centrifugation of citrated whole blood and resuspended in Tyrode’s buffer (2 × 108/mL). aPL (purified IgG from APS patients, 100 μg/mL) was added to induce aggregation. Groups included: aPL only, aPL + PEG hydrogel (pre-incubated), and aPL + αIIbβ3 antibody (10 μg/mL).
Aggregation was measured by turbidimetry (Chrono-log aggregometer), and maximal aggregation (%) was calculated relative to the positive control (10 μM ADP).
2.4. Fibrin clot formation kinetics
Platelet-poor plasma was mixed with thrombin (0.5 U/mL) and CaCl2 (10 mM) to initiate clotting. aPL and corresponding interventions (PEG hydrogel or αIIbβ3 antibody) were added to 96-well plates. OD405 was recorded every 5 min at 37°C for 30 min using a plate reader to monitor fibrin formation.
2.5. Cytokine release from trophoblasts
HTR-8/SVneo cells (1 × 105 cells/well) were plated in 24-well plates. After 24 h, culture media were replaced with platelet-conditioned supernatants from different treatment groups. After 24 h co-incubation, supernatants were collected and cytokine concentrations were quantified using ELISA kits (R&D Systems, validated for cell culture supernatants). Data were normalized to cell counts and expressed as pg per 105 trophoblast cells. Preliminary control assays confirmed the applicability of these kits to platelet-conditioned media.
2.6. Transwell migration assay
HTR-8/SVneo cells (5 × 104 cells/well) were seeded in serum-free medium in the upper chambers of 24-well transwell inserts (8 μm pores, Corning). The lower chambers contained 600 μL of platelet-conditioned medium. After 24 h, migrated cells were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet, and counted in five random fields per insert under an inverted microscope.
All experiments were performed with at least three independent replicates. Data are presented as mean ± standard deviation (SD). Statistical analyses were conducted using OriginPro 2022 (OriginLab Corporation, Northampton, MA, USA). Comparisons between two groups were evaluated using unpaired two-tailed Student’s t-tests. Data are presented as mean ± SD (n = 3). Statistical significance was determined using one-way ANOVA with post hoc Tukey’s test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant.
As depicted in Figure 1, the pathophysiological process begins with circulating antiphospholipid antibodies (aPL) interacting with platelet surfaces, triggering platelet activation. Upon activation, platelets upregulate integrin αIIbβ3 expression, a key receptor responsible for inter-platelet binding via fibrinogen bridges. This enhanced adhesion leads to platelet aggregation and ultimately thrombosis within placental blood vessels. The schematic highlights the role of integrin αIIbβ3 as a central mediator of aPL-driven thrombotic pathology in the placenta.
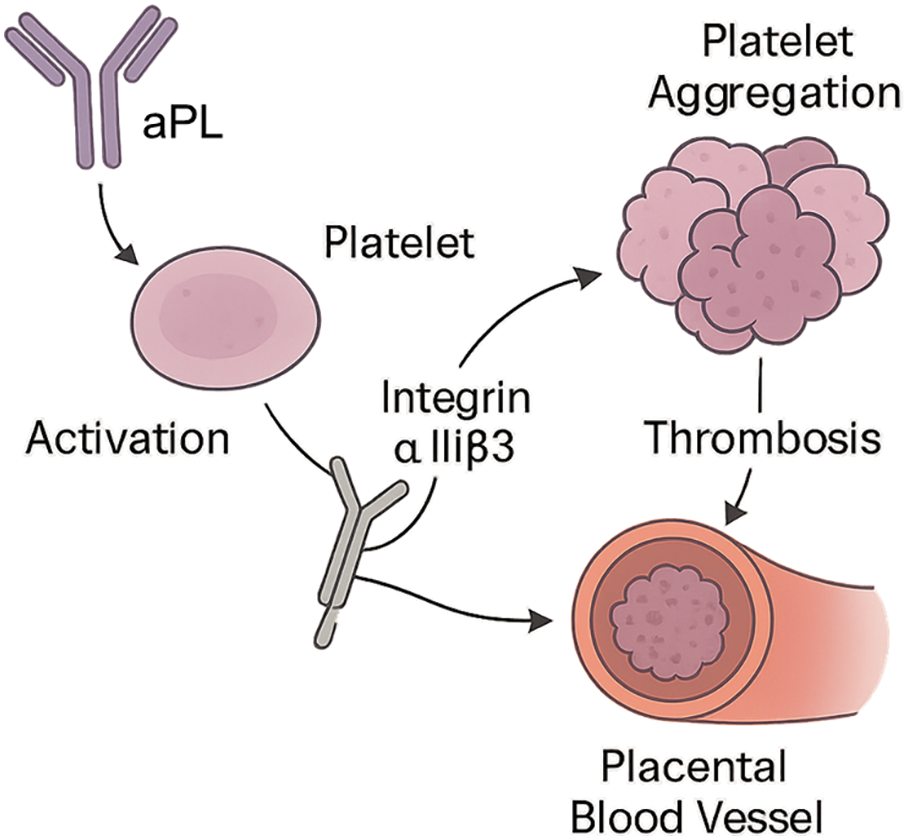
Figure 1. Schematic illustration of aPL-induced platelet activation and integrin αIIbβ3-mediated placental thrombosis
As presented in Figure 2, integrin αIIbβ3 binding exhibited a strong positive correlation with the density of RGD ligands within the PEGDA hydrogel matrix. The 0 μmol/g group, representing the PEGDA hydrogel without RGD modification, served as the baseline control and showed only minimal nonspecific binding (OD450 ~0.1). In contrast, increasing RGD concentrations (10, 50, and 100 μmol/g) led to progressively higher integrin binding, confirming a dose-dependent relationship. When RGD density was increased to 10 μmol/g, the OD450 rose to approximately 0.35. Further increasing the density to 50 μmol/g and 100 μmol/g resulted in OD450 values of ~0.75 and ~0.95, respectively. These results indicate a clear dose-dependent enhancement of αIIbβ3 binding affinity in response to increased RGD functionalization of the hydrogel.
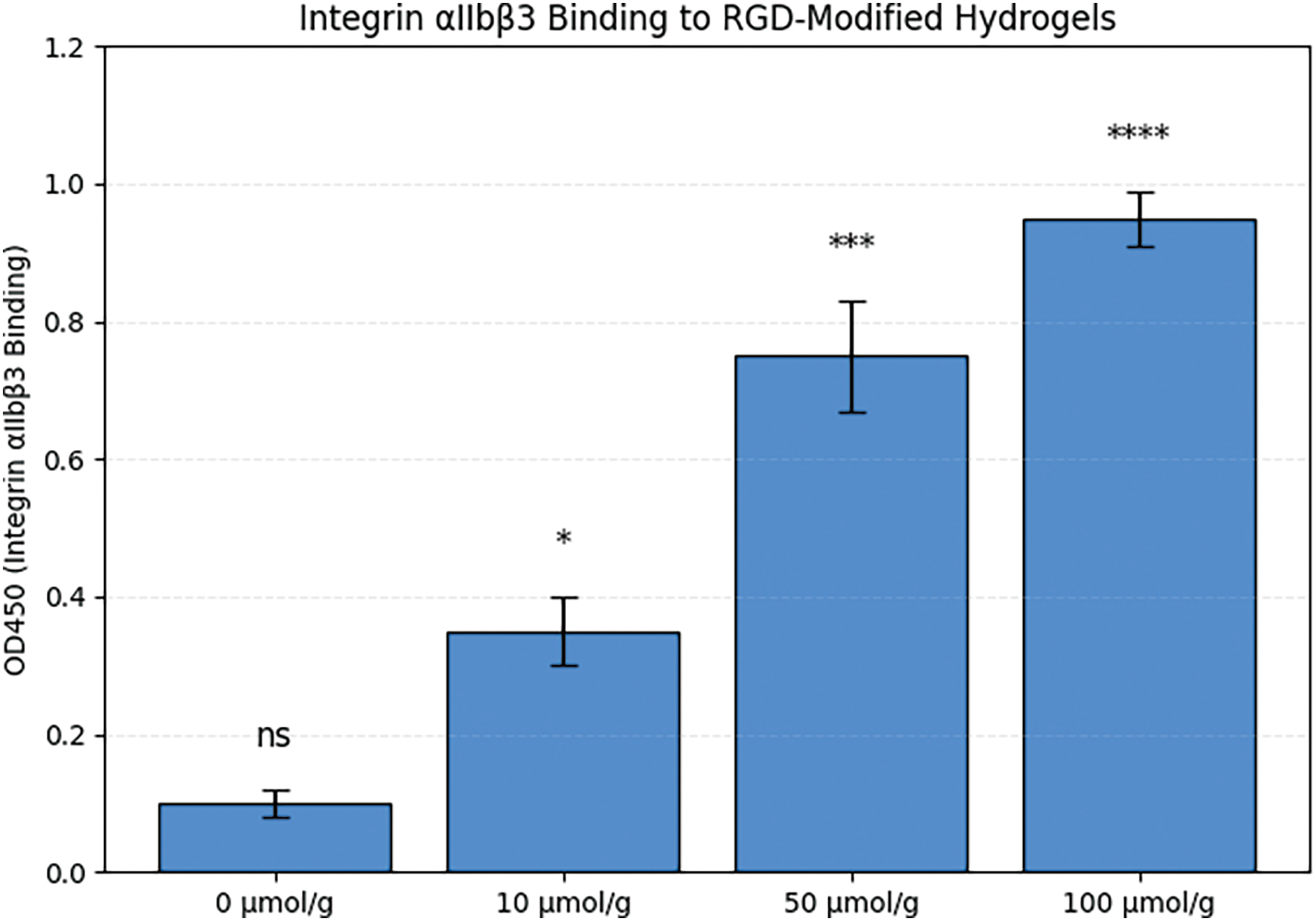
Figure 2. Binding affinity of integrin αIIbβ3 to PEGDA-RGD hydrogels with varying RGD densities. Note. *p < 0.05, ***p < 0.001, ****p < 0.0001; ns, not significant.
As shown in Figure 3, aPL stimulation significantly enhanced platelet aggregation, reaching approximately 90%, compared to a minimal baseline aggregation (~5%) observed in the control group. Upon introduction of the PEG hydrogel, the aggregation rate decreased to ~60%, indicating a partial but notable inhibitory effect. More strikingly, treatment with a specific integrin αIIbβ3 antibody further reduced platelet aggregation to approximately 15%, suggesting superior blockade of the platelet–platelet adhesion pathway. These findings demonstrate that both biomaterial and molecular-level interventions are effective in mitigating the pro-thrombotic effects of aPL.
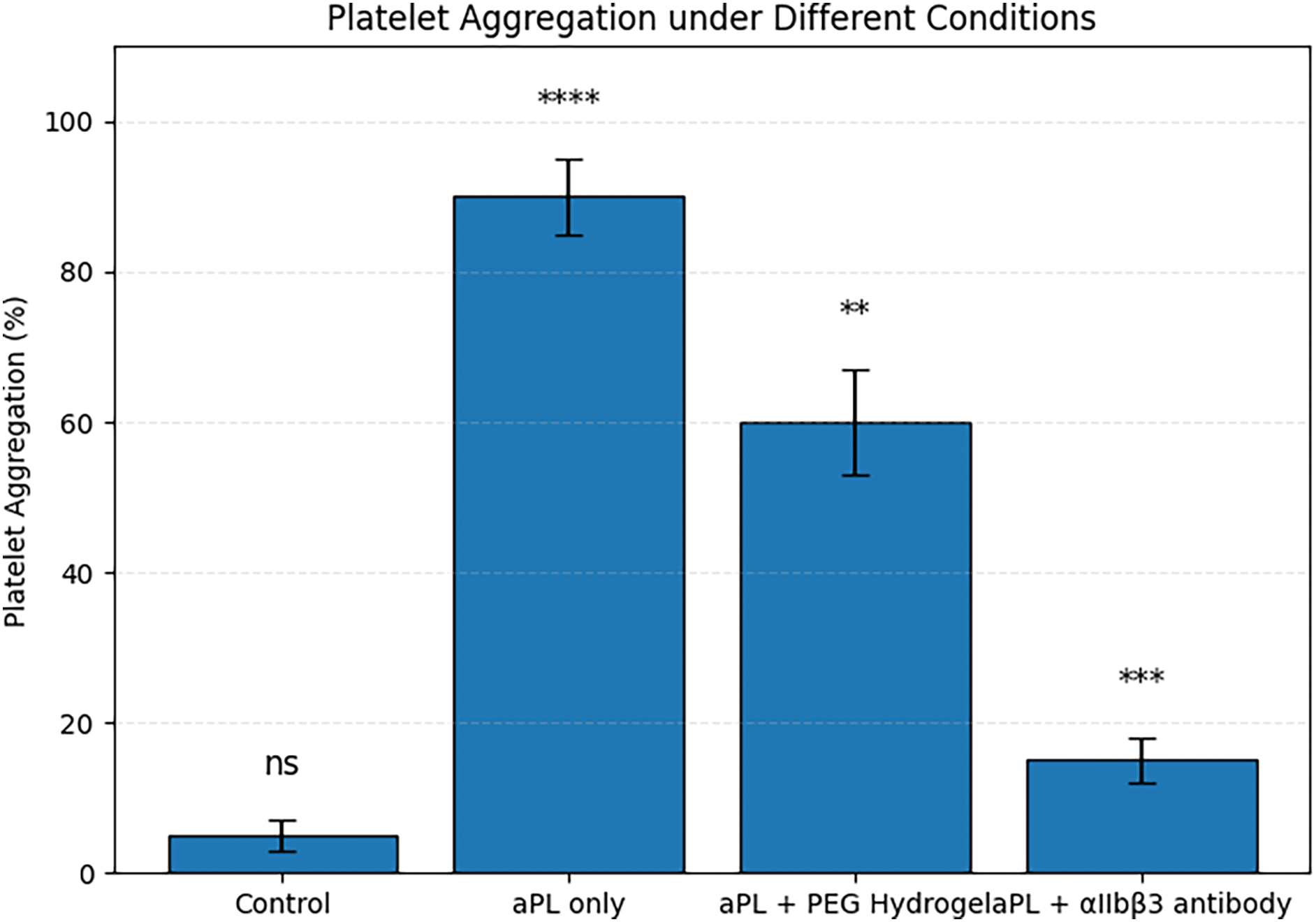
Figure 3. Platelet aggregation in response to antiphospholipid antibody (aPL) stimulation and material-based interventions. Note. **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant.
As shown in Figure 4, the aPL-only group exhibited a rapid and pronounced increase in OD405, peaking around 1.8 by 25 min, indicating accelerated fibrin clot formation. In contrast, the control group without aPL stimulation showed a slower increase, reaching a plateau around 1.1. The group treated with aPL and PEG hydrogel demonstrated intermediate clotting kinetics, with OD405 rising more gradually and reaching ~1.3 by 25 min. Notably, the group treated with aPL and αIIbβ3 antibody showed markedly reduced OD405 throughout the entire time course, with a final value of only ~0.6, reflecting significantly suppressed fibrin polymerization.
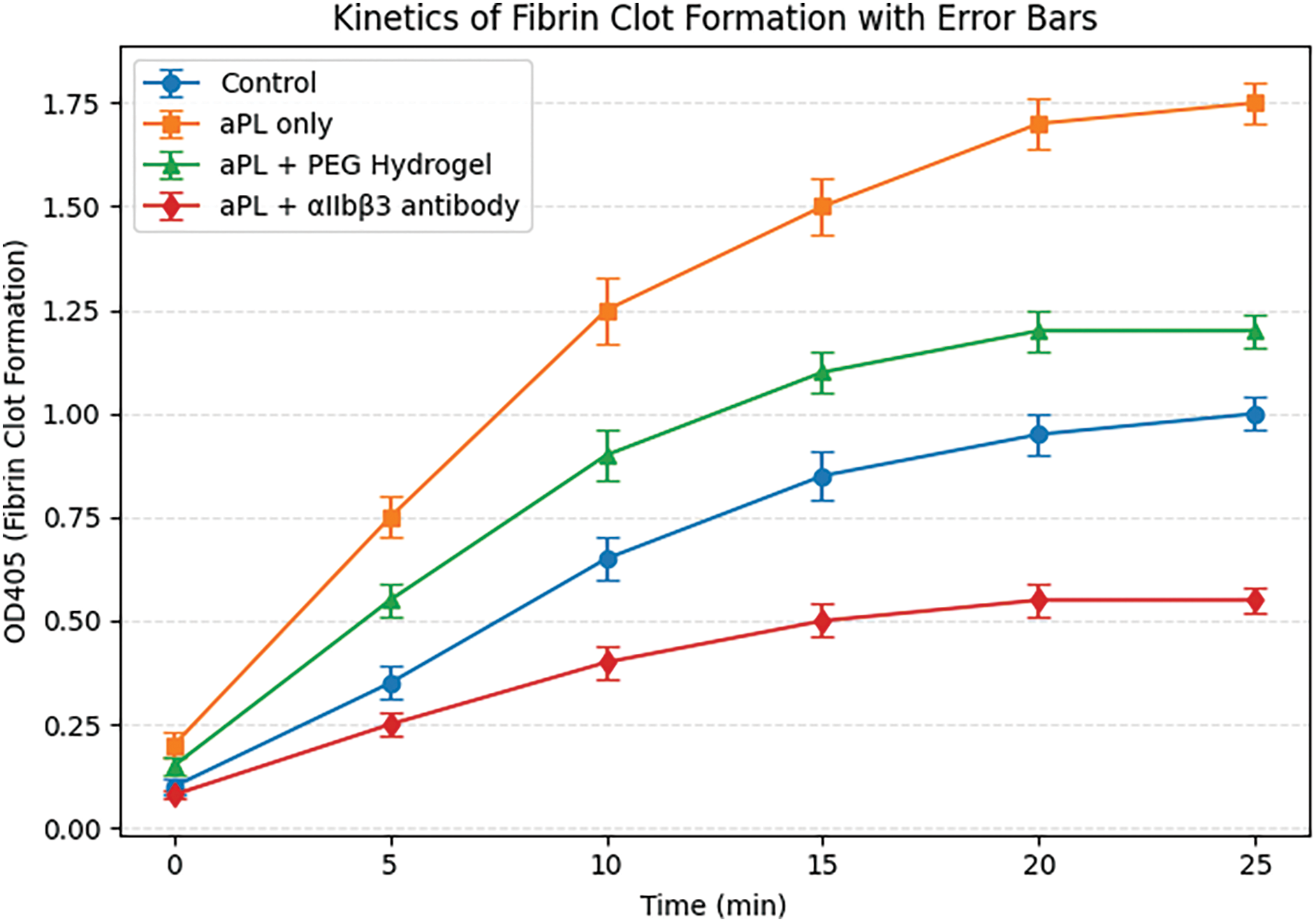
Figure 4. Time-dependent fibrin clot formation in response to aPL and biomaterial interventions
As illustrated in Figure 5, the control group showed low baseline cytokine levels (IL-6 ~20 pg/mL, TNF-α ~10 pg/mL). Upon aPL stimulation, IL-6 and TNF-α levels increased dramatically to ~150 and ~120 pg/mL, respectively, indicating a robust inflammatory response likely mediated by platelet activation. When platelet supernatant from the aPL + PEG hydrogel group was applied, cytokine release was partially attenuated (IL-6 ~100 pg/mL, TNF-α ~85 pg/mL). In contrast, the aPL + αIIbβ3 antibody group demonstrated a more pronounced reduction in cytokine levels (IL-6 ~35 pg/mL, TNF-α ~25 pg/mL), approaching control values.
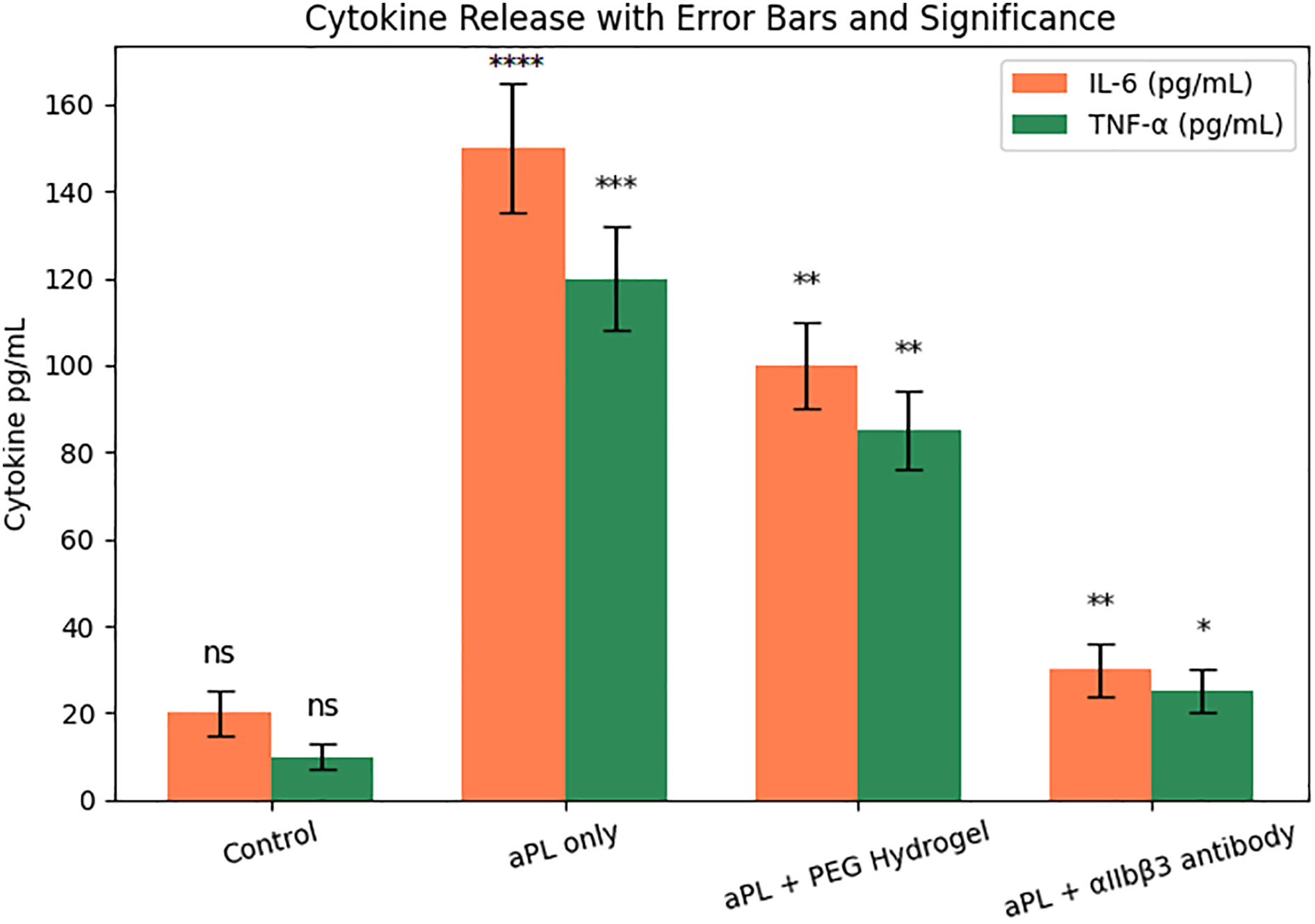
Figure 5. Pro-inflammatory cytokine release from trophoblasts exposed to aPL and platelet-conditioned media with or without material-based interventions. Note. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant.
As shown in Figure 6, the number of migrated trophoblast cells under control conditions reached ~220 cells/field, indicating high basal motility. However, exposure to aPL-stimulated platelet-conditioned media significantly suppressed migration to ~80 cells/field, suggesting strong inhibition of trophoblast motility. In the aPL + PEG hydrogel group, migration was moderately improved (~140 cells/field), while the aPL + αIIbβ3 antibody group nearly restored migration to control levels (~200 cells/field). These results demonstrate that both interventions can alleviate aPL-induced migratory suppression, with the antibody exhibiting greater efficacy.
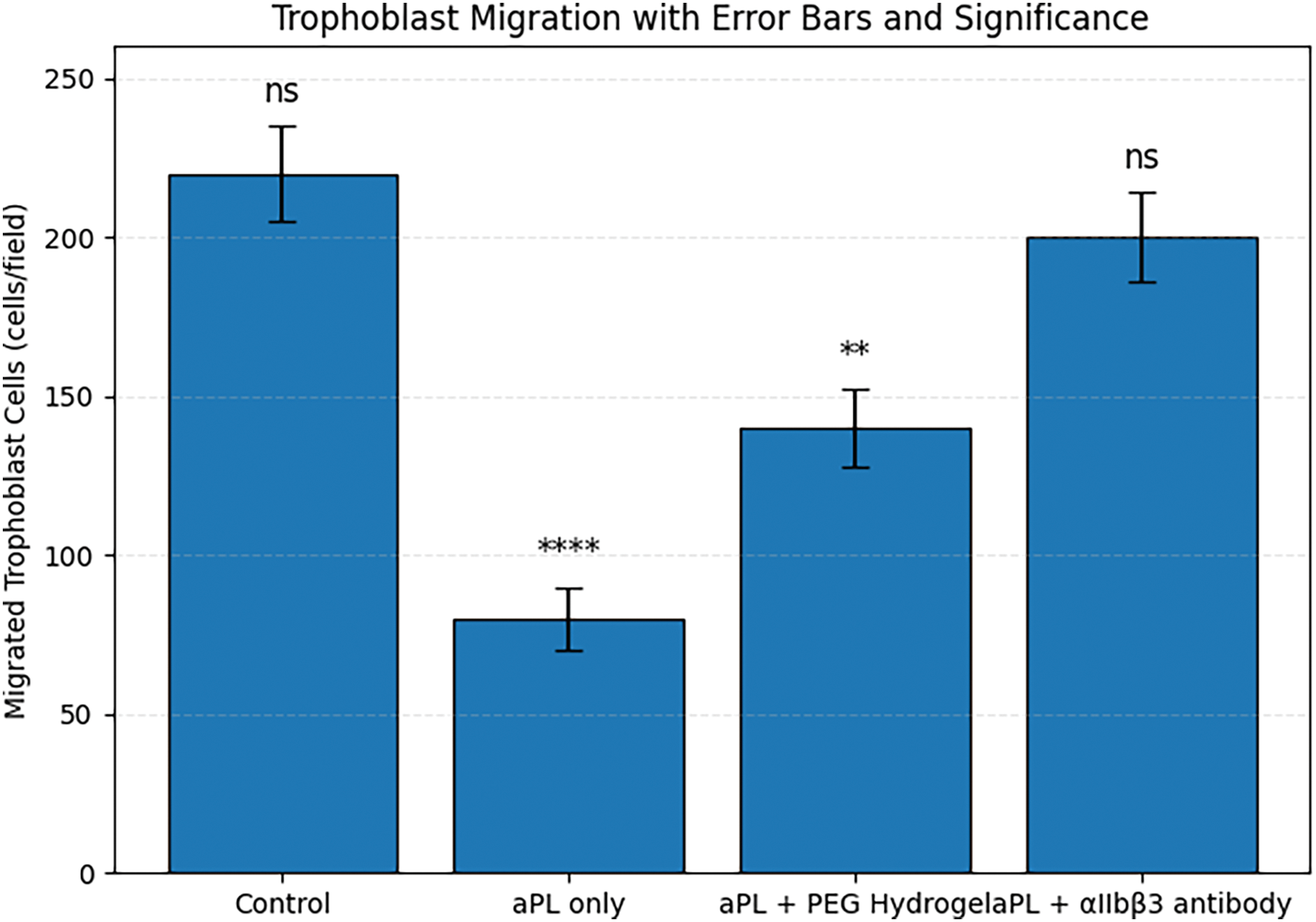
Figure 6. Trophoblast cell migration in response to aPL and biomaterial-based interventions. **p < 0.01, ****p < 0.0001; ns, not significant
As shown in Figure 7A,B, both hydrogels exhibited a porous 3D network, but the RGD-modified hydrogel demonstrated a more compact and interwoven pore architecture. This morphological difference may enhance water diffusion and molecular transport. Figure 7C shows that the RGD-modified hydrogel reached swelling equilibrium within 3 h, with a maximum swelling ratio of ~1.5. This indicates rapid hydration and a well-developed hydrophilic network structure. In Figure 7D, the hydrogel maintained over 99.5% of its mass across a 14-day incubation in PBS, indicating negligible degradation and excellent long-term stability under physiological conditions.
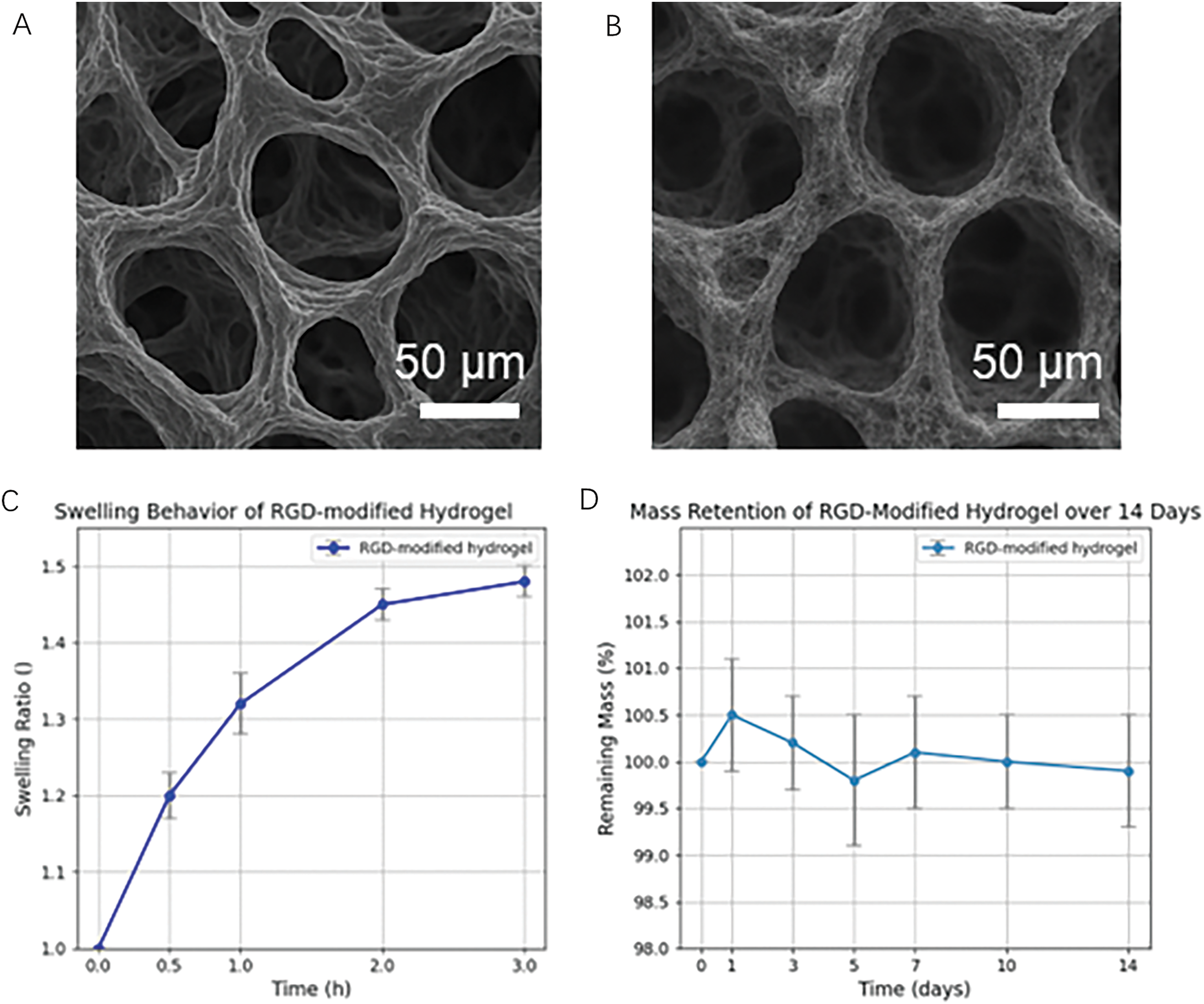
Figure 7. Characterization of the RGD-modified hydrogel (A,B) SEM images of hydrogels with (A) and without (B) RGD modification. Both samples show a porous network structure, with the RGD-modified hydrogel exhibiting slightly denser and more interconnected pores. Scale bar: 50 μm. (C) Swelling behavior of RGD-modified hydrogel within 3 h, reaching equilibrium at a swelling ratio of approximately 1.5. (D) Mass retention of the RGD-modified hydrogel over 14 days in PBS at 37°C, showing stable structure with no significant mass loss
Previous studies have demonstrated the anti-thrombosis potential of RGD-functionalized biomaterials. Kumar et al. showed that RGD-modified peptide grafts reduce platelet adhesion while enhancing endothelialization [23]. Similarly, RGD motifs have been recognized for their ability to inhibit platelet aggregation by blocking integrin engagement. Hydrogels themselves have also been engineered to prevent platelet adhesion; for example, agarose- and gelatin-based hydrogel coatings were found to significantly reduce surface-induced platelet activation [24]. However, these approaches have not been tested in the context of autoimmune thrombophilia. In the present study, we show that PEGDA-based RGD hydrogels competitively inhibit integrin αIIbβ3-mediated platelet aggregation, delay fibrin formation, and attenuate trophoblast dysfunction in a model mimicking antiphospholipid antibody-induced thrombosis. Figure 1 provides a mechanistic overview of how aPL can initiate thrombotic events in pregnancy. The aim of this study was to determine whether RGD-functionalized PEGDA hydrogels can competitively inhibit integrin αIIbβ3 and thereby mitigate the pathogenic effects of antiphospholipid antibodies. Our results demonstrate that the RGD hydrogels effectively bound integrin αIIbβ3 in a dose-dependent manner, significantly reduced platelet aggregation, and delayed fibrin clot formation. Furthermore, the hydrogel intervention suppressed inflammatory cytokine release from trophoblasts and restored their migration capacity. These findings support the concept that biomaterial-based strategies can target integrin αIIbβ3 to attenuate both thrombotic and inflammatory pathways relevant to antiphospholipid syndrome.
Figure 2 underscores the importance of ligand density in governing integrin–material interactions. The αIIbβ3 integrin is central to platelet aggregation through its recognition of RGD motifs in fibrinogen and other adhesive proteins. By systematically increasing RGD content in the PEGDA hydrogel, the material becomes progressively more effective at engaging this receptor [25]. From a biomedical engineering perspective, this dose-dependent binding profile offers valuable design flexibility. At moderate RGD densities (e.g., 10–50 μmol/g), the hydrogel may act as a competitive decoy, partially sequestering αIIbβ3 and blunting platelet activation. At higher densities (100 μmol/g), near-saturation of integrin binding is observed, which may be useful in strongly anti-aggregatory applications, such as localized prevention of aPL-induced thrombosis in pregnancy. Thus, Figure 2 provides mechanistic evidence that functionalizing biomaterials with tunable RGD levels can modulate integrin engagement, supporting their rational design for therapeutic anti-thrombotic interventions.
Figure 3 provides experimental validation for the therapeutic potential of biomaterials targeting platelet aggregation in the context of aPL-related pathology. The near-complete aggregation seen in the “aPL only” group reflects the potent pro-aggregatory activity of antiphospholipid antibodies through activation of integrin αIIbβ3. PEG hydrogels, though inert in composition, appear to reduce aggregation—possibly by sterically hindering platelet–platelet interactions or altering local surface properties. PEG hydrogels are inherently non-fouling and cell-nonadhesive. The hydrated PEG network forms a steric/hydration layer that minimizes nonspecific adsorption of key adhesive proteins (e.g., fibrinogen and vWF), thereby limiting platelet adhesion and activation on the material surface. This physicochemical barrier explains the partial reduction of aPL-induced aggregation observed for the PEG hydrogel group.
In contrast, the αIIbβ3-targeting antibody showed stronger suppression of aggregation, consistent with direct and specific inhibition of the receptor–ligand interaction [26]. These results underscore the central role of integrin αIIbβ3 in aPL-induced thrombosis and highlight the feasibility of using materials-based or molecular therapeutics to interrupt this pathway. In a clinical context, such strategies may offer localized anti-thrombotic protection without systemic bleeding risks, which is especially important in pregnancy-associated APS [27,28].
Figure 4 highlights the thrombogenic potential of antiphospholipid antibodies (aPL) and the protective effects of targeted interventions. aPL-induced platelet activation leads to release of procoagulant factors and accelerates thrombin generation and fibrin formation, as reflected by the sharp increase in OD405 in the aPL-only group. PEG hydrogel appears to attenuate this process, possibly by spatially hindering platelet–fibrin interactions or modulating the local coagulation environment. The most effective suppression was observed in the αIIbβ3 antibody group, which likely results from direct inhibition of platelet aggregation, reducing the procoagulant surface area necessary for fibrin assembly. These findings demonstrate that both material-based and molecular interventions can delay and reduce thrombus formation, supporting their potential utility in managing aPL-mediated placental thrombosis.
Figure 5 provides functional evidence of how aPL-induced platelet activation can exacerbate trophoblast inflammation through the release of paracrine mediators. The significant upregulation of IL-6 and TNF-α upon exposure to aPL-stimulated platelet-conditioned media underscores the crosstalk between coagulation and immune responses in the context of antiphospholipid syndrome (APS).
The observed mitigation of cytokine levels by PEG hydrogel treatment suggests that even inert biomaterials can partially disrupt the activation cascade, possibly by dampening platelet activation or reducing proinflammatory mediator release [29]. Notably, the αIIbβ3 antibody exerted a stronger inhibitory effect, likely due to its direct blockade of integrin-mediated platelet aggregation and degranulation. These results support a mechanistic link between platelet activation and trophoblast inflammation and highlight the potential of integrin-targeted interventions to prevent cytokine-driven placental dysfunction in APS [30].
Figure 6 provides functional evidence that aPL-induced platelet activation negatively impacts trophoblast migration, a key process in successful placentation. Trophoblast motility is essential for vascular remodeling and maternal–fetal interface maintenance, and its disruption is associated with pregnancy complications such as preeclampsia and fetal growth restriction [31]. The reduced migration observed in the aPL-only group is consistent with prior evidence linking platelet-derived pro-inflammatory and pro-thrombotic signals to impaired trophoblast behavior. Treatment with PEG hydrogel partially rescued cell motility, likely by attenuating the inflammatory and thrombogenic environment [32]. More impressively, αIIbβ3 antibody treatment nearly normalized migration, suggesting that integrin-mediated platelet aggregation plays a central role in mediating these downstream effects [33,34]. Together, Figure 6 underscores the therapeutic relevance of blocking platelet aggregation—particularly via integrin αIIbβ3—in preserving trophoblast function under aPL challenge, offering a potential strategy for reducing APS-related placental dysfunction.
This study has certain limitations. First, all experiments were performed in vitro using platelet preparations and trophoblast cell lines, and the findings may not fully recapitulate the complexity of in vivo placental thrombosis. Second, we primarily focused on integrin αIIbβ3 as the therapeutic target, while other signaling pathways involved in antiphospholipid syndrome were not addressed. Third, only two pro-inflammatory cytokines (IL-6 and TNF-α) were analyzed, which provides a partial view of the immune response. Finally, the PEGDA-RGD hydrogel formulation used here was tested under simplified laboratory conditions, and its degradation behavior, long-term stability, and safety remain to be evaluated in vivo. Future work will aim to address these limitations by extending to animal models and exploring additional molecular markers.
The denser pore structure observed in the RGD-modified hydrogel (Figure 7A) may result from additional intermolecular interactions introduced by the peptide, leading to enhanced physical crosslinking and network compaction. This microstructural adjustment correlates with the rapid yet limited swelling observed (Figure 7C), making the hydrogel suitable for applications requiring dimensional stability after hydration. Notably, the hydrogel retained nearly 100% of its mass during the 14-day incubation (Figure 7D), confirming its robust stability in aqueous environments. This is particularly advantageous for long-term biomedical applications such as wound healing matrices or injectable scaffolds, where persistent structure and function are required. The introduction of RGD motifs further suggests potential improvements in cellular affinity and bioactivity.
This study demonstrates that antiphospholipid antibodies (aPL) trigger platelet activation and integrin αIIbβ3-mediated aggregation, leading to thrombosis and inflammatory responses that compromise trophoblast function. By employing PEG-based biomaterials functionalized with RGD peptides, or using αIIbβ3-targeting antibodies, we successfully attenuated aPL-induced platelet aggregation, suppressed fibrin clot formation, and reduced the release of pro-inflammatory cytokines such as IL-6 and TNF-α. Importantly, these interventions also preserved trophoblast migration capacity, which is essential for placental vascular remodeling. Together, our findings suggest that integrin-focused biomaterial strategies may offer a promising localized intervention strategy to mitigate aPL-associated pregnancy complications by combining anti-thrombotic and anti-inflammatory effects.
Acknowledgement: Not applicable.
Funding Statement: The authors received no specific funding for this study.
Author Contributions: Nan Sheng and Yi Tang contributed equally to this work, including conceptualization, methodology, data analysis, and manuscript drafting. Jiahui Qian and Jingwen Xu assisted in validation and data curation. Yunzhao Xu supervised the study, provided project administration, and secured funding. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data that support the findings of this study are available from the Corresponding Author, [Yunzhao Xu], upon reasonable request.
Ethics Approval: Not applicable.
Conflicts of Interest: The authors declare no conflicts of interest to report regarding the present study.
How to Cite this Article
References
- Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, et al. Antiphospholipid syndrome. Nat Rev Dis Primers. 2018;4(1):17103. doi:10.1038/nrdp.2017.103; 29321641 DOI
- Arachchillage DJ, Pericleous C. Evolution of antiphospholipid syndrome. Semin Thromb Hemost. 2023;49(3):295–304. doi:10.1055/s-0042-1760333; 36646109 DOI
- Andreoli L, Regola F, Caproli A, Crisafulli F, Fredi M, Lazzaroni MG, et al. Pregnancy in antiphospholipid syndrome: what should a rheumatologist know? Rheumatology. 2024;63(SI):SI86–95. doi:10.1093/rheumatology/kead537; 38320595 DOI
- Arachchillage DJ, Laffan M, Pericleous C. Hydroxychloroquine as an immunomodulatory and antithrombotic treatment in antiphospholipid syndrome. Int J Mol Sci. 2023;24(2):1331. doi:10.3390/ijms24021331; 36674847 DOI
- Grygiel-Górniak B, Mazurkiewicz L. Positive antiphospholipid antibodies: observation or treatment? J Thromb Thrombolysis. 2023;56(2):301–14. doi:10.1007/s11239-023-02834-6; 37264223 DOI
- Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, de Ramón E, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheumatic Dis. 2015;74(6):1011–8.
- Lewis K, Tambralli A, Madison JA. Pediatric antiphospholipid syndrome: expanding our understanding of antiphospholipid syndrome in children. Curr Opin Rheumatol. 2025;37(3):176–84. doi:10.1097/bor.0000000000001083; 39981610 DOI
- Ding ZT, Pan HY, Yang ZX, Yang CD, Shi H. Beyond the classics: the emerging value of anti-phosphatidylserine/prothrombin antibodies in antiphospholipid syndrome. Clin Immunol. 2023;256:109804. doi:10.1016/j.clim.2023.109804; 37838215 DOI
- Li JY, Ding QL, Wang H, Wu ZX, Gui XC, Li CW, et al. Engineering smart composite hydrogels for wearable disease monitoring. Nano-Micro Lett. 2023;15(1):105. doi:10.1007/s40820-023-01079-5; 37060483 DOI
- Gaspar P, Sciascia S, Tektonidou MG. Epidemiology of antiphospholipid syndrome: macro- and microvascular manifestations. Rheumatology. 2024;63(SI):SI24–36. doi:10.1093/rheumatology/kead571; 38320589 DOI
- Gao D, Sun CW, Woodley AB, Dong JF. Clot retraction and its correlation with the function of platelet integrin αIIbβ3. Biomedicines. 2023;11(9):2345. doi:10.3390/biomedicines11092345; 37760786 DOI
- Dodig S, Cepelak I. Antiphospholipid antibodies in patients with antiphospholipid syndrome. Biochemia Medica. 2024;34(2):020504. doi:10.11613/bm.2024.020504; 38882589 DOI
- Devreese KMJ. Noncriteria antiphospholipid antibodies in antiphospholipid syndrome. Int J Lab Hematol. 2024;46(S1):34–42. doi:10.1111/ijlh.14268; 38584293 DOI
- del-Pino RA, Monahan RC, Huizinga TWJ, Eikenboom J, Steup-Beekman GM. Risk factors for antiphospholipid antibodies and antiphospholipid syndrome. Semin Thromb Hemost. 2024;50(6):817–28. doi:10.1055/s-0043-1776910; 38228166 DOI
- Chae A, Murali G, Lee SY, Gwak J, Kim SJ, Jeong YJ, et al. Highly oxidation-resistant and self-healable MXene-based hydrogels for wearable strain sensor. Adv Funct Mater. 2023;33(24):2213382.
- Feng WJ, Wang ZK. Tailoring the swelling-shrinkable behavior of hydrogels for biomedical applications. Adv Sci. 2023;10(28):2303326. doi:10.1002/advs.202303326; 37544909 DOI
- Li XY, Gong JP. Design principles for strong and tough hydrogels. Nat Rev Mater. 2024;9(6):380–98. doi:10.1038/s41578-024-00672-3. DOI
- de Assis V, Giugni CS, Ros ST. Evaluation of recurrent pregnancy loss. Obstet Gynecol. 2024;143(5):645–59. doi:10.1097/aog.0000000000005498; 38176012 DOI
- Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Investig. 2003;112(11):1644–54. doi:10.1172/jci200318817. DOI
- Situ AJ, Ulmer TS. Comparison of integrin αIIbβ3 transmembrane association in vesicles and bicelles. Biochemistry. 2023;62(12):1858–63. doi:10.1021/acs.biochem.3c00177; 37279176 DOI
- Grossi C, Capitani N, Benagiano M, Baldari CT, Della Bella C, Macor P, et al. Beta 2 glycoprotein I and neutrophil extracellular traps: potential bridge between innate and adaptive immunity in anti-phospholipid syndrome. Front Immunol. 2023;13:1076167. doi:10.3389/fimmu.2022.1076167; 36700193 DOI
- Li YJ, Yang D, Wu ZY, Gao FL, Gao XZ, Zhao HY, et al. Self-adhesive, self-healing, biocompatible and conductive polyacrylamide nanocomposite hydrogels for reliable strain and pressure sensors. Nano Energy. 2023;109(17):108324. doi:10.1016/j.nanoen.2023.108324. DOI
- Kumar VB, Tiwari OS, Finkelstein-Zuta G, Rencus-Lazar S, Gazit E. Design of functional RGD peptide-based biomaterials for tissue engineering. Pharmaceutics. 2023;15(2):345. doi:10.3390/pharmaceutics15020345; 36839667 DOI
- Apte G, Lindenbauer A, Schemberg J, Rothe H, Nguyen T-H. Controlling surface-induced platelet activation by agarose and gelatin-based hydrogel films. ACS Omega. 2021;6(16):10963–74. doi:10.1021/acsomega.1c00764; 34056249 DOI
- Sorice M, Profumo E, Capozzi A, Recalchi S, Riitano G, Veroli BDi, et al. Oxidative stress as a regulatory checkpoint in the production of antiphospholipid autoantibodies: the protective role of NRF2 pathway. Biomolecules. 2023;13(8):1221. doi:10.3390/biom13081221; 37627286 DOI
- Zou JM, Sun SY, Simone IDe, ten Cate H, de Groot PG, de Laat B, et al. Platelet activation pathways controlling reversible integrin αIIbβ3 activation. TH Open. 2024;8(2):e232–42. doi:10.1055/s-0044-1786987; 38911141 DOI
- Stammler R, Nguyen Y, Yelnik C, Guern VLe, Lambert M, Paule R, et al. Precipitating factors of catastrophic antiphospholipid syndrome: the role of anticoagulant treatment in a series of 112 patients. J Thromb Haemost. 2023;21(5):1258–65. doi:10.1016/j.jtha.2023.02.007; 36792010 DOI
- Soto-Peleteiro A, Gonzalez-Echavarri C, Ruiz-Irastorza G. Obstetric antiphospholipid syndrome. Med Clin. 2024;163:S14–21. doi:10.1016/j.medcli.2024.05.001; 39174149 DOI
- Xue P, Valenzuela C, Ma SS, Zhang X, Ma JZ, Chen YH, et al. Highly conductive MXene/PEDOT:PSS-integrated poly(N-isopropylacrylamide) hydrogels for bioinspired somatosensory soft actuators. Adv Funct Mater. 2023;33(24):2214867. doi:10.1002/adfm.202214867. DOI
- Zhao B, Bai ZY, Lv HL, Yan ZK, Du YQ, Guo XQ, et al. Self-healing liquid metal magnetic hydrogels for smart feedback sensors and high-performance electromagnetic shielding. Nano-Micro Lett. 2023;15(1):79. doi:10.1007/s40820-023-01043-3; 37002442 DOI
- Tektonidou MG. Update on antiphospholipid syndrome. Rheumatology. 2024;63(SI):SI1–3. doi:10.1093/rheumatology/kead633; 38320590 DOI
- Thang NH, Chien TB, Cuong DX. Polymer-based hydrogels applied in drug delivery: an overview. Gels. 2023;9(7):523. doi:10.3390/gels9070523; 37504402 DOI
- Wang ZW, Wei H, Huang YJ, Wei Y, Chen J. Naturally sourced hydrogels: emerging fundamental materials for next-generation healthcare sensing. Chem Soc Rev. 2023;52(9):2992–3034. doi:10.1039/d2cs00813k; 37017633 DOI
- Xin HL, Huang JS, Song ZQ, Mao JH, Xi XD, Shi XF. Structure, signal transduction, activation, and inhibition of integrin αIIbβ3. Thromb J. 2023;21(1):18. doi:10.1186/s12959-023-00463-w; 36782235 DOI